SOYEZ CONSCIENTS QUE CE SONT DES MALADIES RARES : LES DIAGNOSTICS TARDIFS ET DIFFÉRENTIELS SONT FRÉQUENTS AVEC LES MALADIES NEUROMUSCULAIRES
On m’a diagnostiqué une dystrophie musculaire après des années d’examens et de visites chez les médecins, mais il y avait encore quelque chose qui clochait. J’ai continué de pousser pour avoir un diagnostic précis et je suis contente de l’avoir fait, car j’ai finalement un nom pour ma maladie.
– Ana*
Un diagnostic tardif est fréquent
Il y a des retards de diagnostic pour les maladies neuromusculaires, souvent de plusieurs années. Même des maladies héréditaires présentes, voire symptomatiques à la naissance, sont souvent diagnostiquées des décennies plus tard1.
Dans le cas de la maladie de Pompe, par exemple, l’âge moyen au moment du diagnostic chez les adultes est la mi-trentaine, alors que les symptômes apparaissent souvent 10 ans avant le diagnostic2. Pour les patients qui ont une maladie neuromusculaire traitable, ce temps perdu est particulièrement préoccupant.
Le diagnostic tardif s’explique par l’apparition discrète, sur plusieurs années, de symptômes qui ressemblent à une fatigue généralisée et par les similitudes et chevauchements du tableau clinique avec celui d’autres maladies neuromusculaires3. C’est pourquoi en documentant une anamnèse approfondie et des évaluations cliniques et en faisant une orientation rapide vers le bon spécialiste, vous pouvez faire gagner un temps précieux à votre patient1.
La maladie de Pompe, l’une des quelque 7 000 maladies rares, toucherait environ 1 personne sur 40 000, mais sa prévalence réelle en Amérique du Nord est inconnue4,5.
Une détection précoce change tout
Lorsqu’un patient souffre de faiblesse musculaire et de symptômes associés – parfois depuis des années – et ignore ce qui les cause, le seul fait d’avoir un diagnostic peut être un soulagement. Le patient peut ainsi avoir un nom pour son problème, et mieux savoir comment gérer sa vie dès lors et préparer son avenir. Un diagnostic peut aussi lui permettre d’obtenir un traitement et de tenter d’améliorer la prise en charge de ses symptômes.
Mettre l’accent sur le diagnostic
De nombreux troubles qui se manifestent par une faiblesse et une atrophie musculaires, dont la dystrophie musculaire, les myopathies inflammatoires, l’amyotrophie musculaire spinale et la maladie de Pompe, ont des symptômes similaires. On compte des dizaines de types de dystrophie musculaire, qui supposent tous une éventuelle perte de force musculaire, une incapacité croissante et de possibles déformations des articulations (contractures, scoliose, etc.)6.
Il existe deux principaux groupes de maladies musculaires7 :
- Troubles musculaires qui causent la destruction des fibres musculaires, entraînant (souvent de façon progressive) une faiblesse et une fonte musculaires
- Maladies qui causent plus de défaillances fonctionnelles que de dégénérescence des fibres structurales (peu de fonte) telles que myopathies métaboliques ou canalopathies
Les maladies qui détruisent progressivement les fibres musculaires se divisent en deux grandes catégories7 :
- Dystrophies : maladies dégénératives non inflammatoires du muscle qui, en général, sont génétiquement déterminées, non curables efficacement et évolutives
- Myopathies inflammatoires : considérées comme étant auto-immunes dans la plupart des cas
Retour en haut de la page
Diagnostics différentiels pour les maladies neuromusculaires
Les tableaux cliniques semblables des différentes maladies neuromusculaires peuvent compliquer leur diagnostic différentiel. Il est important de connaître ces maladies afin de réduire le plus possible les retards de diagnostic et d’accélérer l’établissement d’un diagnostic précis et la prise en charge pour les patients.
Voici quelques maladies neuromusculaires qui ont des signes et symptômes communs3,6,8–24 :
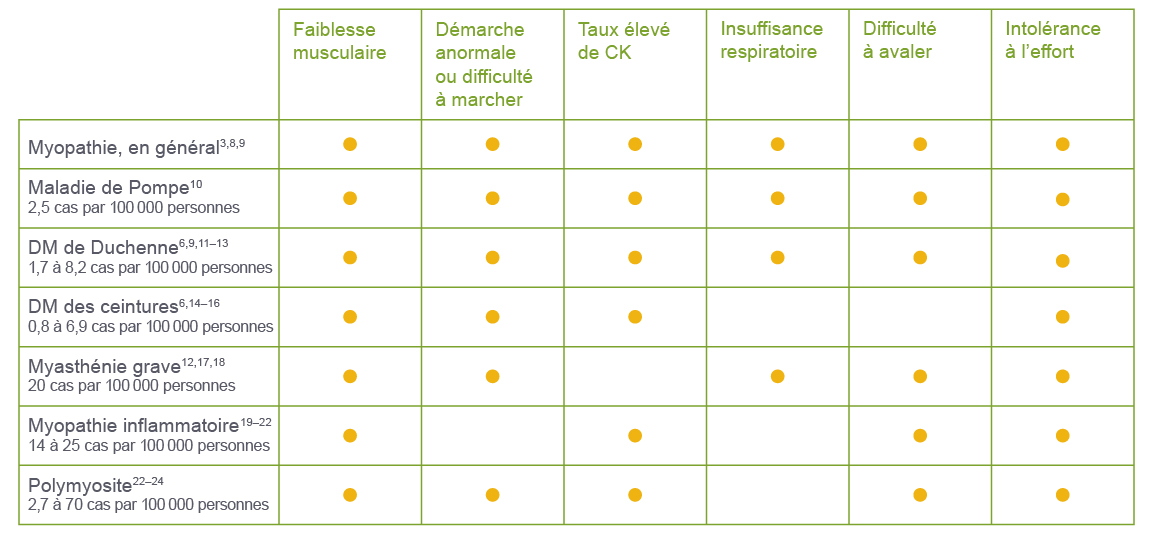
La liste des signes et symptômes énumérés dans ce tableau n’est pas exhaustive et ne comprend pas tous les signes et symptômes qui pourraient survenir chez une personne atteinte de ces maladies.
Références
Références : 1. Grigull L, et al. Diagnostic support for selected neuromuscular diseases using answer-pattern recognition and data mining techniques: a proof of concept multicenter prospective trial. BMC Medical Informatics and Decision Making 2016;16. doi: 10.1186/s12911-016-0268-5. 2. Toscano A, Montagnese F et Musumeci O. Early is better? A new algorithm for early diagnosis in Late Onset Pompe Disease (LOPD). Acta Myol 2013;32(2):78–81. Accessible à l’adresse : https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3866896/. 3. Barohn RJ, et al. A pattern recognition approach to the patient with a suspected myopathy. Neurol Clin 2014;32(3):569–vii. Accessible à l’adresse : https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4233647/. 4. Kishnani PS, et al. Late-Onset Pompe Disease: Presentation, Diagnosis, and Management - A Continuing Medical Education Monograph. AANEM. Accessible à l’adresse : https://www.aanem.org/mxonline/resources/downloads/products/ED01.pdf. 5. National Organization for Rare Disorders (NORD). Accessible à l’adresse : http://www.rarediseases.org. 6. Barnabei MS, et al. Exercise and muscular dystrophy: implications and analysis of effects on musculoskeletal and cardiovascular systems. Compr Physiol 2011;1(3):1353–63. 7. Reeves et Swenson. Neuromuscular system disorders. Dans: Disorders of the nervous system. Year Book Medical Pub; 1980. 8. Hereditary myopathy with early respiratory failure. Genetics Home Reference website. Accessible à l’adresse : https://ghr.nlm.nih.gov/condition/hereditary-myopathy-with-early-respiratory-failure. Publié le 21 février 2017. Consulté le 27 février 2017. 9. Jaradeh S. GI Motility Online website. Accessible à l’adresse : http://www.nature.com/gimo/contents/pt1/full/gimo35.html. Consulté le 28 juillet 2020. 10. Kishnani PS, et al. Pompe disease diagnosis and management guideline. Genet Med 2006;8(5):267–88. 11. Mah JK, et al. A systematic review and meta-analysis on the epidemiology of Duchenne and Becker muscular dystrophy. Neuromuscul Disord 2014;24(6):482–91. 12. Gilchrist JM. Overview of neuromuscular disorders affecting respiratory function. Semin Respir Crit Care Med 2002;23(3):191–200. 13. Ozawa E, Hagiwara Y et Yoshida M. Creatine kinase, cell membrane and Duchenne muscular dystrophy. Mol Cell Biochem 1999;190(1–2):143–51. 14. Limb-girdle muscular dystrophy. Genetics Home Reference website. Accessible à l’adresse : https://ghr.nlm.nih.gov/condition/limb-girdle-muscular-dystrophy. Publié le 21 février 2017. Consulté le 27 février 2017. 15. Pegoraro E et Hoffman EP. Limb-girdle muscular dystrophy overview. Dans : Pagon RA, Adam MP, Ardginger HH, et al., eds. GeneReviews. Seattle, Washington: University of Washington, Seattle; 2014. Accessible à l’adresse : https://www.ncbi.nlm.nih.gov/books/NBK1408. Mis à jour le 30 août 2012. Consulté le 1er mars 2017. 16. Siciliano G, et al. Acta Myol 2015;34(1):3–8. 17. Myasthenia gravis. Genetics Home Reference website. Accessible à l’adresse : https://ghr.nlm.nih.gov/condition/ myasthenia-gravis. Publié le 21 février 2017. Consulté le 27 février 2017. 18. Myasthenia gravis: what is it? Harvard Health Publishing website. Accessible à l’adresse : https://www.health.harvard.edu/a_to_z/myasthenia-gravis-a-to-z. Consulté le 29 juillet 2020. 19. Smoyer-Tomic KE, Amato AA et Fernandes AW. Incidence and prevalence of idiopathic inflammatory myopathies among commercially insured, Medicare supplemental insured, and Medicaid enrolled populations: an administrative claims analysis. BMC Musculoskelet Disord 2012;13:103. doi: 10.1186/1471-2474-13-103. 20. Furst D, Amato A et Iorga Ş. Epidemiology of adult idiopathic inflammatory myopathies in a U.S. managed care plan. Muscle & Nerve 2012;45:676–83. 21. NINDS inflammatory myopathies information page. National Institute of Neurological Disorders and Stroke website. Accessible à l’adresse : https://www.ninds.nih.gov/Disorders/All-Disorders/Inflammatory-Myopathies-Information-Page. Consulté le 29 juillet 2020. 22. Mastaglia FL, Phillips BA. Idiopathic inflammatory myopathies: epidemiology, classification, and diagnostic criteria. Rheum Dis Clin North Am 2002;28(4):723–41. 23. Bernatsky S, Joseph L et Pineau CA. Estimating the prevalence of polymyositis and dermatomyositis from administrative data: age, sex and regional differences. Ann Rheum Dis 2009;68:1192–96. 24. Polymyositis. John Hopkins Medicine website. Accessible à l’adresse : https://www.hopkinsmedicine.org/health/conditions-and-diseases/polymyositis. Consulté le 29 juillet 2020.
Retour en haut de la page