BE RARE DISEASE AWARE: DELAYED AND DIFFERENTIAL DIAGNOSES ARE COMMON WITH NEUROMUSCULAR DISEASES
I was diagnosed with muscular dystrophy after years of tests and doctors’ visits, but something still didn’t feel right. I kept pushing for a specific diagnosis and I’m glad I did, because I finally have a name for my disease.
– Ana*
Delayed diagnosis is common
Diagnosis of neuromuscular diseases is delayed, often by many years. Even inherited diseases that are present, if not symptomatic at birth, often take decades to be diagnosed.1
In the case of Pompe disease, for instance, mean age at diagnosis for adults is mid 30s, with onset of symptoms often appearing 10 years before the diagnosis.2 For patients who have a treatable neuromuscular disease, this lost time is particularly concerning.
Delayed diagnosis occurs because of the subtle onset of symptoms over many years that mimic generalized fatigue and due to similarities and overlaps in clinical presentation with other neuromuscular diseases.3 This is why both documenting a thorough history and clinical assessments and making a prompt referral to the appropriate specialist can help save your patient valuable time.1
As one of about 7,000 rare diseases, Pompe disease affects an estimated 1 in 40,000 people, although its true prevalence in North America is unknown.4,5
Early detection makes a difference
When a patient has been suffering with muscle weakness and related symptoms – sometimes for years – and doesn’t know what is causing them, just having a diagnosis can be a relief. It gives the patient a name for their problem, and an idea of how to manage their life moving forward and prepare for the future. It may also allow them to seek treatment and work to manage their symptoms more effectively.
Focusing on diagnosis
Many disorders that present with muscle weakness and atrophy, including muscular dystrophy, inflammatory myopathies, spinal muscular atrophy and Pompe disease, have similar symptoms.
There are many dozens of types of muscular dystrophy – each one involves an eventual loss of muscle strength, increasing disability and possible joint deformity (contractures, scoliosis, etc.).6
Two major groups of muscle disease include:7
- Muscle disorders that cause destruction of muscle fibers, leading to (usually progressive) muscle weakness and wasting
- Diseases that cause more of a functional defect than structural fiber degeneration (little wasting) such as metabolic myopathies or channelopathies
Diseases that progressively destroy muscle fibers fall into two main types:7
- Dystrophies: non-inflammatory degenerative conditions of muscle that are genetically determined, not effectively curable, and progressive
- Inflammatory myopathies: thought to be autoimmune in most cases
Back to top
Differential diagnoses in neuromuscular disease
Overlapping clinical presentations among neuromuscular diseases can make their differential diagnosis challenging. It is important to be aware of these diseases to help minimize diagnostic delays and expedite accurate diagnoses and management for patients.
Here are some neuromuscular diseases that may have shared signs and symptoms:3,6,8–24
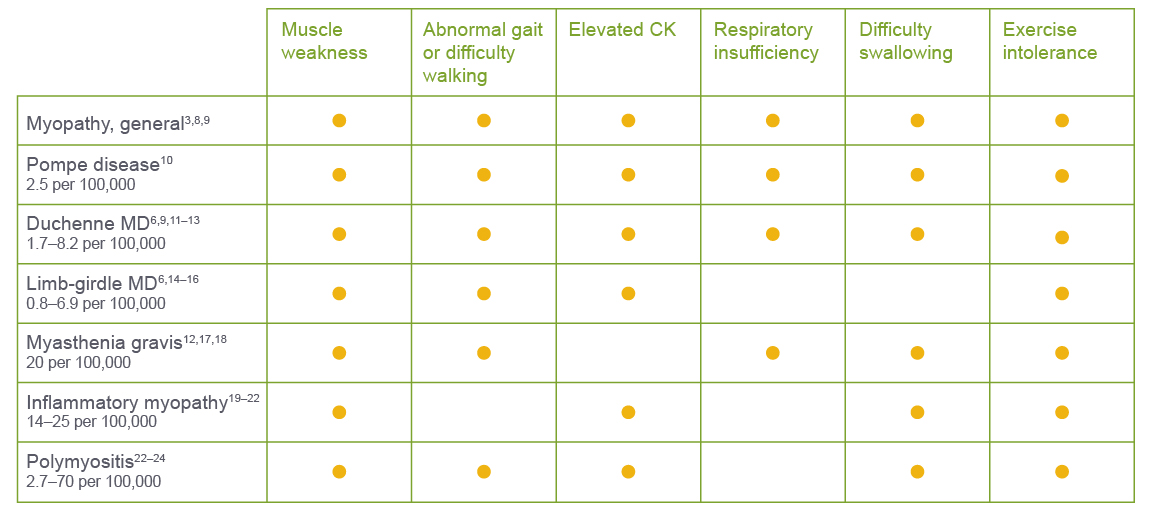
The signs and symptoms listed in this chart are not an exhaustive list and may not be inclusive of all signs and symptoms that someone with these diseases may encounter.
References
References: 1. Grigull L, et al. Diagnostic support for selected neuromuscular diseases using answer-pattern recognition and data mining techniques: a proof of concept multicenter prospective trial. BMC Medical Informatics and Decision Making 2016;16. doi: 10.1186/s12911-016-0268-5. 2. Toscano A, Montagnese F and Musumeci O. Early is better? A new algorithm for early diagnosis in Late Onset Pompe Disease (LOPD). Acta Myol 2013;32(2):78–81. Available at: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3866896/. 3. Barohn RJ, et al. A pattern recognition approach to the patient with a suspected myopathy. Neurol Clin 2014;32(3):569–vii. Available at: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4233647/. 4. Kishnani PS, et al. Late-Onset Pompe Disease: Presentation, Diagnosis, and Management - A Continuing Medical Education Monograph. AANEM. Available at: https://www.aanem.org/mxonline/resources/downloads/products/ED01.pdf. 5. National Organization for Rare Disorders (NORD). Available at: http://www.rarediseases.org. 6. Barnabei MS, et al. Exercise and muscular dystrophy: implications and analysis of effects on musculoskeletal and cardiovascular systems. Compr Physiol 2011;1(3):1353–63. 7. Reeves and Swenson. Neuromuscular system disorders. In: Disorders of the nervous system. Year Book Medical Pub; 1980. 8. Hereditary myopathy with early respiratory failure. Genetics Home Reference website. Available at: https://medlineplus.gov/genetics/condition/hereditary-myopathy-with-early-respiratory-failure/. Published February 21, 2017. Accessed February 27, 2017. 9. Jaradeh S. GI Motility Online website. Available at: https://www.nature.com/gimo/contents/pt1/full/gimo35.html. Accessed July 28, 2020. 10. Kishnani PS, et al. Pompe disease diagnosis and management guideline. Genet Med 2006;8(5):267–88. 11. Mah JK, et al. A systematic review and meta-analysis on the epidemiology of Duchenne and Becker muscular dystrophy. Neuromuscul Disord 2014;24(6):482–91. 12. Gilchrist JM. Overview of neuromuscular disorders affecting respiratory function. Semin Respir Crit Care Med 2002;23(3):191–200. 13. Ozawa E, Hagiwara Y and Yoshida M. Creatine kinase, cell membrane and Duchenne muscular dystrophy. Mol Cell Biochem 1999;190(1–2):143–51. 14. Limb-girdle muscular dystrophy. Genetics Home Reference website. https://ghr.nlm.nih.gov/condition/limb-girdle-muscular-dystrophy. Published February 21, 2017. Accessed February 27, 2017. 15. Pegoraro E and Hoffman EP. Limb-girdle muscular dystrophy overview. In: Pagon RA, Adam MP, Ardginger HH, et al, eds. GeneReviews. Seattle, Washington: University of Washington, Seattle; 2014. Available at: https://www.ncbi.nlm.nih.gov/books/NBK1408. Updated August 30, 2012. Accessed March 1, 2017. 16. Siciliano G, et al. Acta Myol 2015;34(1):3–8. 17. Myasthenia gravis. Genetics Home Reference website. https://ghr.nlm.nih.gov/condition/myasthenia-gravis. Published February 21, 2017. Accessed February 27, 2017. 18. Myasthenia gravis: what is it? Harvard Health Publishing website. Available at: https://www.health.harvard.edu/a_to_z/myasthenia-gravis-a-to-z. Accessed July 29, 2020. 19. Smoyer-Tomic KE, Amato AA and Fernandes AW. Incidence and prevalence of idiopathic inflammatory myopathies among commercially insured, Medicare supplemental insured, and Medicaid enrolled populations: an administrative claims analysis. BMC Musculoskelet Disord 2012;13:103. doi: 10.1186/1471-2474-13-103. 20. Furst D, Amato A and Iorga Ş. Epidemiology of adult idiopathic inflammatory myopathies in a U.S. managed care plan. Muscle & Nerve 2012;45:676–83. 21. Inflammatory myopathies information page. National Institute of Neurological Disorders and Stroke website. Available at: https://www.ninds.nih.gov/Disorders/All-Disorders/Inflammatory-Myopathies-Information-Page. Accessed July 29, 2020. 22. Mastaglia FL, Phillips BA. Idiopathic inflammatory myopathies: epidemiology, classification, and diagnostic criteria. Rheum Dis Clin North Am 2002;28(4):723–41. 23. Bernatsky S, Joseph L and Pineau CA. Estimating the prevalence of polymyositis and dermatomyositis from administrative data: age, sex and regional differences. Ann Rheum Dis 2009;68:1192–96. 24. Polymyositis. Johns Hopkins Medicine website. Available at: https://www.hopkinsmedicine.org/health/conditions-and-diseases/polymyositis. Accessed July 29, 2020.
Back to top